DNA/RNA提取
DNA和RNA不同的提取方案步骤
DNA提取
质粒提取
gDNA提取
RNA提取
RNA提取是分子生物学中的一项基础实验技术,用于从细胞或组织中分离纯化RNA。高质量的RNA是后续如RT-PCR、RNA测序(RNA-seq)、Northern blot等实验的关键
mRNA提取
1.试剂和仪器准备
| Materials | 耗材试剂 | 设备 |
|---|---|---|
2.试剂成分和提取原理
Dynabeads® mRNA DIRECT™ 试剂盒的优势:
• 快速 — 15分钟的程序即可获得纯净、完整的 mRNA
• 高纯 mRNA 分离 — cDNA 合成上游的理想选择
• 灵敏的 mRNA 分离 — 可实现来自超小起始样品的 cDNA 合成和 cDNA 文库构建(支持从单个细胞构建 cDNA 文库)
| Compoent | 容量 |
|---|---|
Dynabeads Ologo(dT)25 (=5mg/mL, supplied in PBS pH7.4) |
5mL |
Lysis/Binding Buffer
|
30mL |
Washing Buffer A
|
60mL |
Washing Buffer B
|
30mL |
| 10mM Tris-HCl Ph 7.5 (Elution Buffer) |
分离方案依赖于在大多数 mRNA 3’ 端 polyA 残基与共价偶联到 Dynabeads® 表面的寡核苷酸 (dT)25 之间的碱基配对。其他缺乏 polyA 尾的 RNA 种类不会与微珠杂交,并且易于洗涤。核糖体 RNA、DNA、蛋白和小 RNA 分子(如转运 RNA、微小 RNA 和小核仁 RNA)不会与珠结合且会被丢弃
1 mg Dynabeads® 寡核苷酸 (dT)25 微珠 (200µL) 能够结合高达 2 µg mRNA。一个常规的哺乳细胞包含10-30pg的total RNA,1%-5%是mRNA
只有当bead和sample比例合适的时候才不会发生mRNA size的bias,当有大量mRNA或孵育时间过短,beads更倾向于短分子
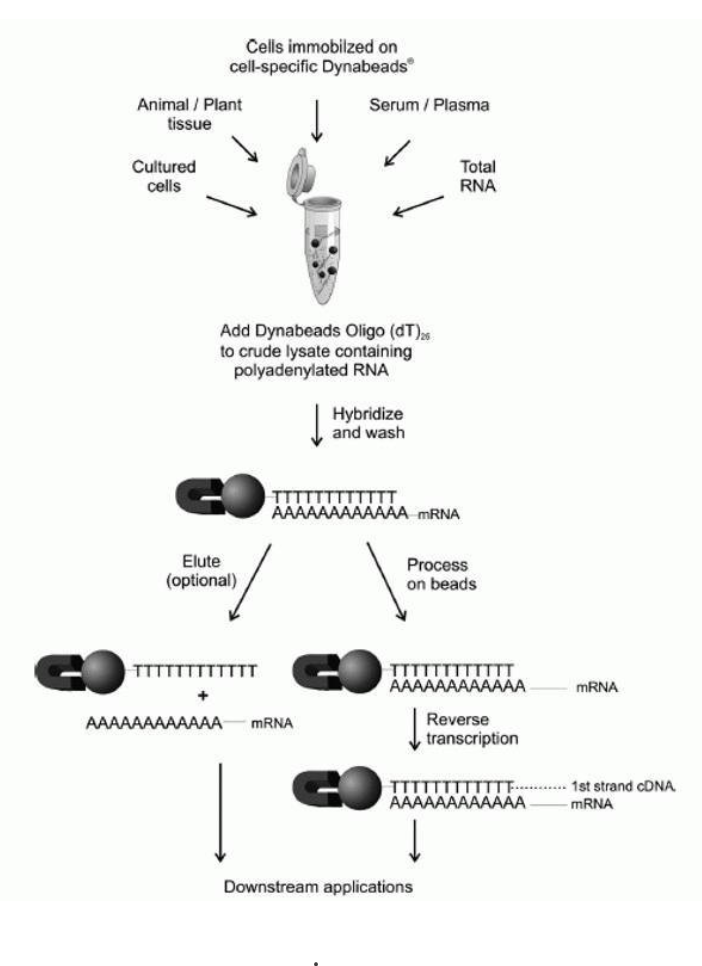
3.步骤
开始前准备
- Beads需要涡旋或者移液器打匀进行重悬,并且在室温回温半小时
- Lysis/Binding Buffer和Washing Buffers A和B提前放置在室温
- 10mM Tris-HCl在使用之前拿出或者放置在2-8度冰浴。
- 需要在wash Beads后彻底吸干净buffer,buffer中含有LiDS–具有强烈的酶活抑制
3.1 样本前处理
植物或动物组织需要液氮速冻后,加入裂解液进行离心,做到之后再进行补充
- 离心收集细胞(400xg,8min,4度)
| 标准量 | Micro | Mini | Maxi |
|---|---|---|---|
| 1-4x106 | <150000 | 0.15-1 x 106 | 4-20 x 106 |
使用PBS/DPBS重悬细胞,重新离心,清洗细胞。
按照不同的细胞量添加不同量的培养基
| 标准量 | Micro | Mini | Maxi |
|---|---|---|---|
| 1250µL | 300µL | 300µL | 5mL |
- 使用移液器多次吹打直到DNA全部释放,得到粘稠的液体
3.2 Dynabeads Oligo(dT)25 准备
- 重悬Beads,按照需要的量将Beads转移到1.5mL无核酶离心管中,将离心管放置到磁力架上
| 标准量 | Micro | Mini | Maxi |
|---|---|---|---|
| 250µL | 10µL | 50µL | 1mL |
- 经过30s或者1min后,当溶液变澄清,移除上清液
- 将离心管从磁力架上移下来,使用表格 Table 1 中的Lysis/Binding Buffer量清洗beads
3.3 mRNA提取
- 将上一步清洗后的beads放置到磁力架上,静置30s或1min,等待beads全部被吸附。
- 移除上清液体后,按照 Table 1 加入合适的样品裂解液,移液器重悬beads
- 将混合后的beads使用HulaMix或者roller mix进行孵育,3-5min,使得mRNA与beads结合
千万不要用涡旋仪进行混匀,会将mRNA打断
- 将孵育后的beads放置到磁力架上2min,如果粘度很多增加到10min左右
- 吸去上清之后,室温下加入合适体积的Wash A Buffer ,重复磁力架-吸取上清的步骤
| 标准量 | Micro | Mini | Maxi |
|---|---|---|---|
| 1-2mL | 600µL | 600µL | 10mL |
- 使用Wash B Buffer 室温下进行清洗,如果分离的mRNA用于后续的酶反应(RT-PCR),再进行额外一次的wash Buffer B 清洗
| 标准量 | Micro | Mini | Maxi |
|---|---|---|---|
| 1-1.5mL | 300µL | 300µL | 5mL |
- mRNA解离,按照表中加入合适的洗脱液10mMTris-HCl pH7.5,然后在65°C–80°C孵育2min,立即将beads放到磁力架上,将Elution Buffer吸取放置到冰上
| 标准量 | Micro | Mini | Maxi |
|---|---|---|---|
| 10-25µL | 10µL | 10µL | 50-100µL |
3.4 rRNA清除(可选)
在一些情况下提取的mRNA会存在rRNA的污染,对于Northern Blot和RT-PCR是没有干扰的,但是对于cDNA文库的构建和microarray分析rRNA需要被避免
原则上是通过重新分离mRNA去洗脱,重新使用beads是被推荐的,否则新的beads需要使用焦磷酸钠清洗
- 按照上述步骤得到洗脱的mRNA
- 将洗脱的mRNA转移到新的无核酶1.5mL离心管中放置在冰上,不要扔掉beads
- 使用Washing Buffer B清洗两次beads
- 使用4倍体积Elution buffer加入Lysis/Binding Bffer(洗脱用20µLbuffer,添加80µL Lysis/Bingding Buffer)
- 移除Washing Buffer B,加入上一步稀释好的mRNA
- 在室温下,孵育3-5min
- 之后进行Washing Buffer A清洗–Washing Buffer B 清洗–洗脱
3.5 RNA定量
试剂Qubit™ RNA 高灵敏度 (HS)、宽范围 (BR) 和扩展范围 (XR) 定量试剂盒 |
样照
|
|
| Qubit3.0 |  {#fig-Qubit width=“300” height=“200” fig-alt = “Qubit2”} {#fig-Qubit width=“300” height=“200” fig-alt = “Qubit2”} |
|
- Qubit RNA HS 定量试剂盒
-
Qubit RNA HS(高灵敏度)定量试剂盒用于准确检测初始浓度为 0.2 至 200 ng/µL 的 RNA 样品(具体取决于样品体积),检测范围为 4−200 ng
- Qubit RNA BR 定量试剂盒
-
Qubit RNA BR(宽范围)定量试剂盒,该定量试剂盒设计用于准确检测初始浓度为 0.5 至 1,200 ng/µL 的 RNA 样品(具体取决于样品量),检测范围为 10−1,200 ng
- Qubit RNA XR 定量试剂盒
-
Qubit RNA XR(扩展范围)定量试剂盒,该定量试剂盒设计用于准确检测初始浓度为 5 至 20,000 ng/µL 的 RNA 样品(具体取决于样品量),检测范围为 100−20,000 ng
- 准备样品和标准品,室温下面回温,最好将染料分装。
- RNA BR assay需要两个标准品进行校准,耗材是特定的0.5mL PCR tubes
- Qubit working solution是 Qubit RNA BR 1:200 稀释在RNA BR Buffer中。
荧光染料,不要在玻璃容器中进行混匀
- 添加标准品或样品到buffer mix中,补足200µL体积
| 标准品测试 | 样本测试 | |
|---|---|---|
| Volume of working solution | 190µL | 180–199 µL |
| Volume of standard | 10 µL | – |
| Volume of user sample | – | 1–20 µL |
| Total volume in each assay tube | 200 µL | 200 µL |
- 涡旋3-5秒
- 室温放置孵育2imn,注意避光。
- 将PCR管放置到Qubit中,在屏幕上点击Home–touch RNA–选择RNA Broad Range
- 按照试剂上的标签,标准品分为#1和#2,先将#1插入Qubit读取,再插入#2插入Qubit读取
- 注意测量方式的选择,不能选错,Qubit分为RNA,dsDNA,ssDNA
- 标准品测定要区分数据
- 将样品放置到Qubit中,选择上样的量(1-20µL),选择合适的单位之后,进行读取
Total RNA提取
1.仪器和试剂准备
| Materials | 耗材试剂 | 设备 |
|---|---|---|
2.试剂成分和提取原理
TRIzol 试剂是一种即用型试剂,用于在 1 小时 内从人类、动物、植物、酵母或细菌来源的细胞和组织样本中提取高质量的总 RNA(以及 DNA 和蛋白质)。TRIzol 试剂含有苯酚、异硫氰酸胍和其他特殊组分的单相溶液,可促进提取各种大或小分子量的 RNA。在 TRIzol 试剂进行样本匀浆化的过程中,它可以破坏细胞,溶解细胞组分,同时高效的抑制 RNA 酶的活性,从而维持 RNA 的完整性。TRIzol 试剂可同时处理大量样本,对 Chomcynski 和 Sacchi 开发的一步法提取 RNA(Chomczynski和 Sacchi,1987)的改进
采用 TRIzol 试剂进行样本匀浆化后,加入氯仿,匀浆物可分成透明的上层水相层(含有 RNA)、相界面和红色的下层有机层(含有 DNA 和蛋白质)。然后用异丙醇从水相层中沉淀出 RNA。用乙醇从相界面和有机层中沉淀出DNA。利用异丙醇沉淀从酚 - 乙醇上清液中沉淀出蛋白质。洗涤沉淀的 RNA、DNA 或蛋白质,去除杂质,重悬浮后供下游应用
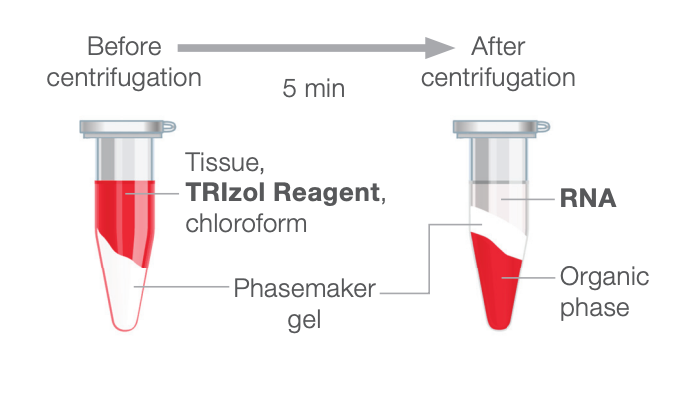
3.核酸提取
3.1 样品前处理
- 组织: 每50-100mg组织(可速冻)加入1mLTRIzol试剂,并用匀浆机破碎
- 单层贴壁细胞
- 移除培养基
- 使用预冷的DPBS/PBS冲洗一遍细胞,吸取干净。有时候为了避免mRNA降解也不进行清洗。
- 加入1mL(可根据细胞量适量减少TRIzol量)TRIzol试剂到培养皿中裂解细胞
- 一定要使用移液器反复吹打使用细胞充分裂解
- 悬浮细胞处理
- 通过离心收集细胞并弃掉上清
- 由于悬浮细胞量大,向细胞中加入1mLTRIzol
- 反复吹打使细胞裂解
- 组织或细胞可以放置在-80°C中保存,直到使用前加入TRIzol
- 也可以加入TRIzol后,样本可以在4°C保存过夜,第二天处理,也可以放在-20°C保存长达一年,-80°C更长时间的保存,标签一定要做好
- TRIzol味道很大,有很强的挥发性,建议在通风橱中进行
3.2 Total RNA提取
- 将上一步裂解的TRIzol混合物移到透明的1.5mL离心管中,室温孵育5min使核蛋白复合物完全解离
- 每1mL的TRIzol试剂中添加0.2mL氯仿,盖紧试管盖子,然后通过摇动彻底混合
氯仿加进去之后,由于密度大,会沉在离心管底部,一定要进行摇匀,否则离心之后会失败
- 室温孵育2-3min
- 在 4° C 下 12,000×g 离心样本 15 分钟
混合物分离成下层的红色苯酚 - 氯仿层和以及无色的上层水相
- 通过45° 倾斜试管并将透明溶液吸出,将含有RNA的水相转移到新管中
主要是吸取透明的上清,不能吸取白色的中间层和下面红色曾,如果吸取了可以打掉只保留透明的部分,45°或者垂直吸取都可以,也可以保留一些不必要全部抽取干净
- 1mLTRIzol大约能抽取0.4-0.5mL的上清液,然后加入0.5mL异丙醇
- 4°C或者室温孵育10min
- 在4°C下12,000×g离心10分钟
总RNA沉淀在试管底部形成白色凝胶状沉淀
- 使用移液器小心的去掉上清液
3.3 洗涤RNA
- 按照每1mLTRIZol试剂裂解处理所得到的沉淀重悬于75%乙醇中
- 短暂涡旋样本后,在在4°C下7500×g离心5分钟
- 使用微量移液器弃去上清液
- 将RNA沉淀在真空或空气下干燥5-10分钟
!切勿用真空离心机干燥沉淀。切勿让RNA沉淀过度干燥。以确保RNA的完全溶解。部分溶解的 RNA样本的A230/280比值<1.6
3.4 溶解测量RNA
- 使用20-50 μL不含RNase的水,或者TE Buffer(官方还要加0.1mMEDTA或0.5%SDS,看自己需求)反复吹打使沉淀重新悬浮
- 在55-60°C的水浴或加热块中孵育10-15分钟
- 立即使用放置在冰浴中,或者长期保存在-70°C下
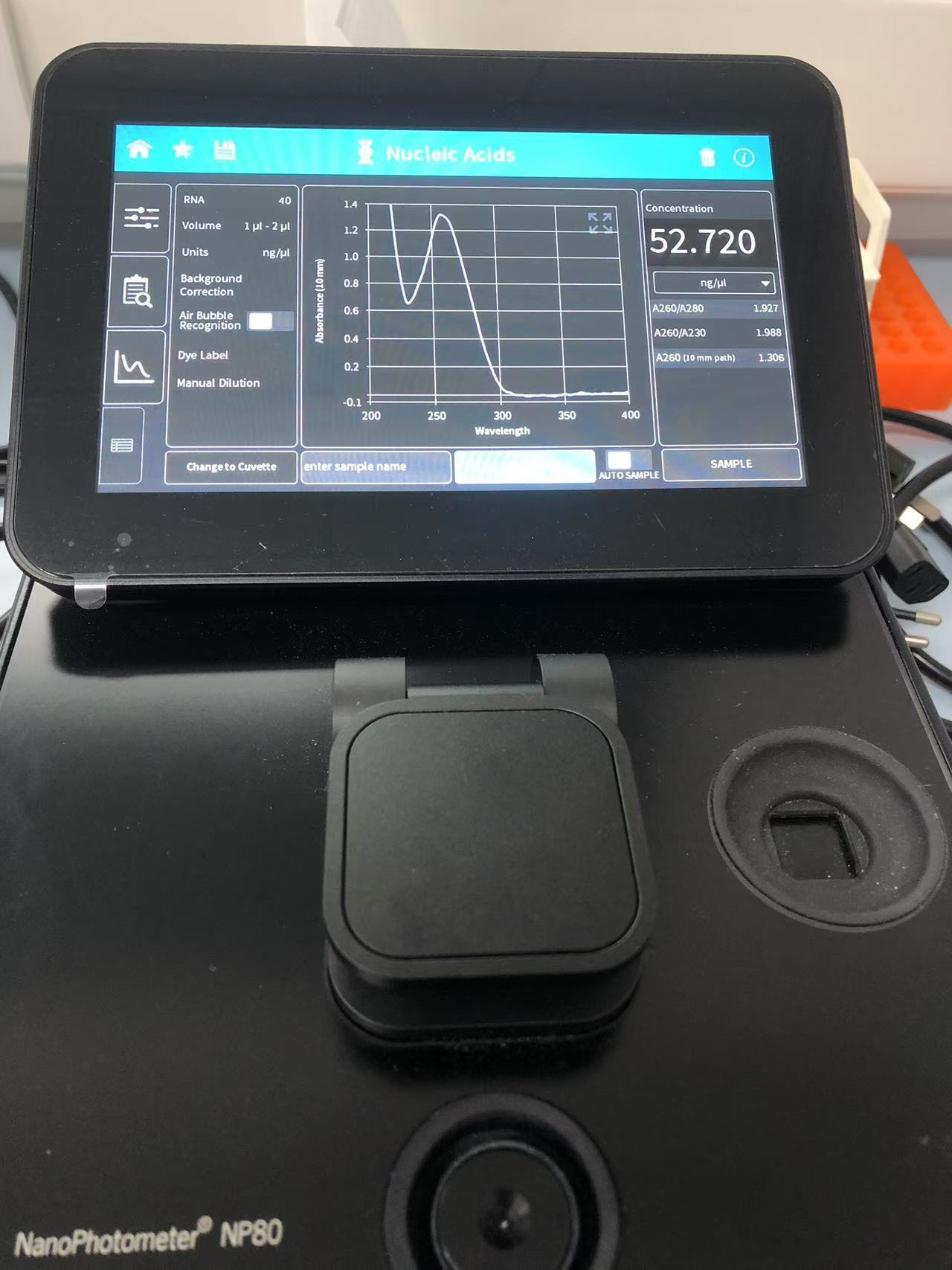
- 测定RNA含量和纯度。
每个仪器操作上可能有细微差别,但是总体一致,得到A260/A280, A260/A230来查看RNA的纯度和含量,更精确的定量,可以使用 Qubit
4. 污染判断
A260表示核酸在最高吸收峰260nm波长处的吸光度值,即260nm出核酸最容易吸收光,通过检测260nm处吸收的吸光度值可评测纯化双链DNA,单链DNA或RNA样品的浓度。
| 波长(nm) | 主要检测物质 | 应用 |
|---|---|---|
| 260 nm | 核酸(RNA、DNA) | 用于计算核酸浓度 |
| 280 nm | 蛋白质(含芳香族氨基酸,如Trp、Tyr) | 用于判断蛋白污染 |
| 230nm | 有机物/盐类(乙醇、GTC、GuHCl、EDTA等) | 用于判断有机污染物 |
| 比值 | 正常值 | 偏离情况 | 可能的污染物或原因 |
|---|---|---|---|
| A260/A280 | ~2.0 | <1.8 | 蛋白质、酚类 |
| A260/A280 | >2.1 | 偏高可能为背景干扰或低浓度误差 | 仪器误差、杂质干扰 |
| A260/A230 | 2.0–2.2 | <1.8 | GTC(异硫氰酸胍)、GuHCl(盐酸胍)、乙醇、EDTA |
| A260/A230 | >2.4 | 不常见,可能为测定误差 | 波长设置问题或杂质吸收峰移位 |
在 Table 2 中就可以得到下面中常见的干扰因素,分母越大比值越小,按照常见污染物的吸收峰即可大概推测到污染物来源,Table 3 只能作为辅助,并不是绝对的。之前还根据A260/A280大于2来判断RNA是否降解,但是仅仅是经验之谈,还需要跑胶验证。
RNA的糖环比DNA多一个自由羟基,并且环境中存在大量的RNA酶,因此提取RNA很容易出现降解的情况。紫外分光光度计方法很难分辨提取的RNA是否完整,因为无论时完整的(intact RNA)还是片段化的RNA(degraded RNA)在260 nm处都会有一个吸光值
EDTA在~230 nm处有最高吸收峰,因此会降低A260/A230比值,但是当EDTA螯合Mg2+或Ca2+后,EDTA-阳离子复合物的紫外吸光度会显著低于游离EDTA,因此在含有二价阳离子的EDTA溶液中测量DNA A260/A230比值很可能超过3.0。这也是为什么pure RNA在10 mM Tris pH 8.5溶液中A260/A230比值在2.3-2.4之间,而在TE缓冲液(Tris-EDTA)中A260/A230比值在2.6-3.0之间
核酸的提取仅仅是下游应用的开始,注意枪头,手,唾液,离心管和水中的核酶污染基本能控制核酸的完整性,希望都能提到纯纯的核酸,顺利进行下一步